Usage
BioNetGen modelling language is a language for writing rule-based models of biochemical systems, including signal transduction, metabolic, and genetic regulatory networks, see here for more information.
This VS Code extension is designed to help write BNGL models by adding syntax highlighting and snippet support, do rapid tests of the model as you write with the help of a built-in run button and basic plotting features.
Syntax highlighting and snippets
Once the extension is installed you can create a new file with .bngl. This file extension
will be automatically detected and you should see a run button at the top right corner of the
file if the extension is running correctly. This extension will also do syntax highlighing on
files with .net extension.
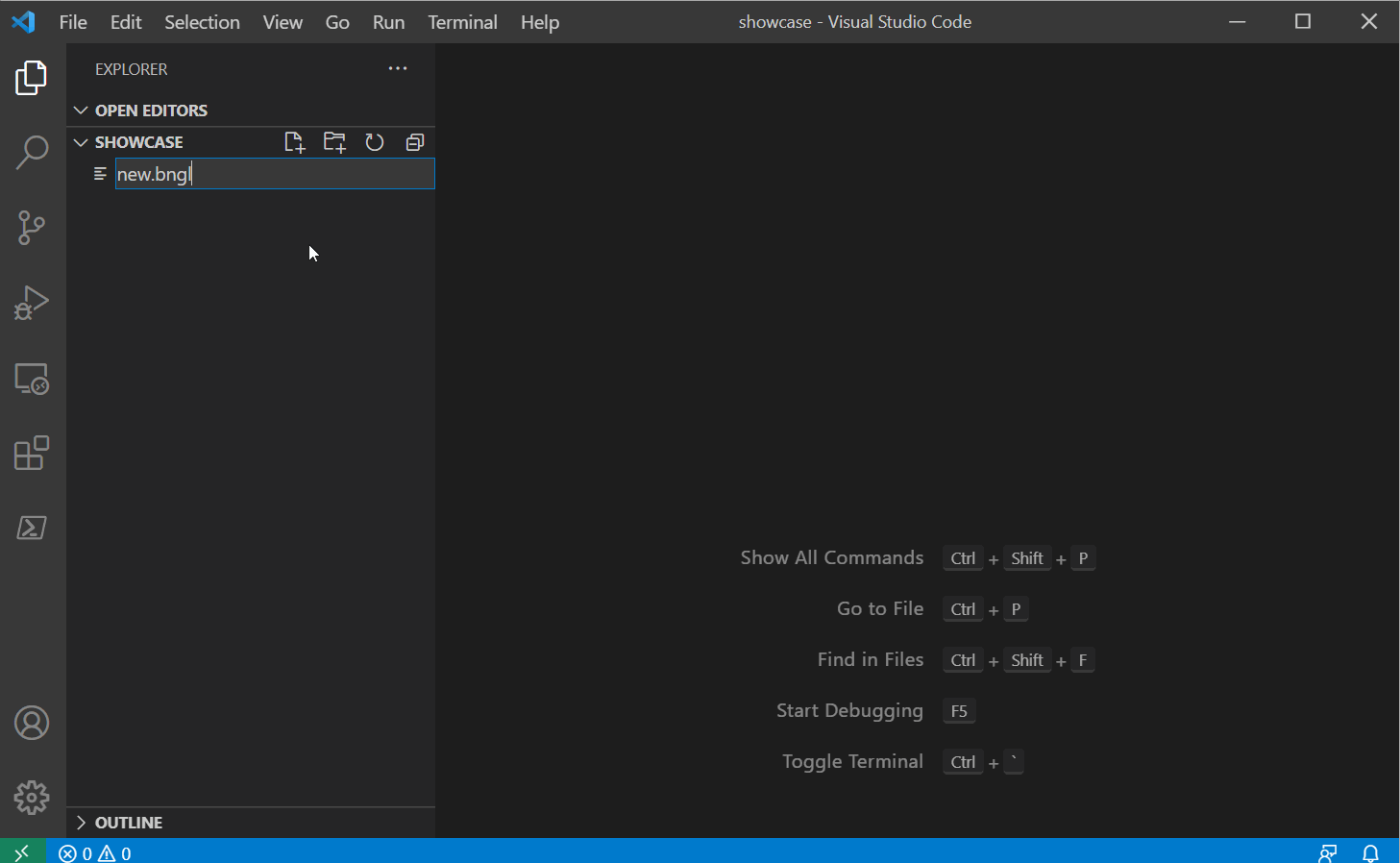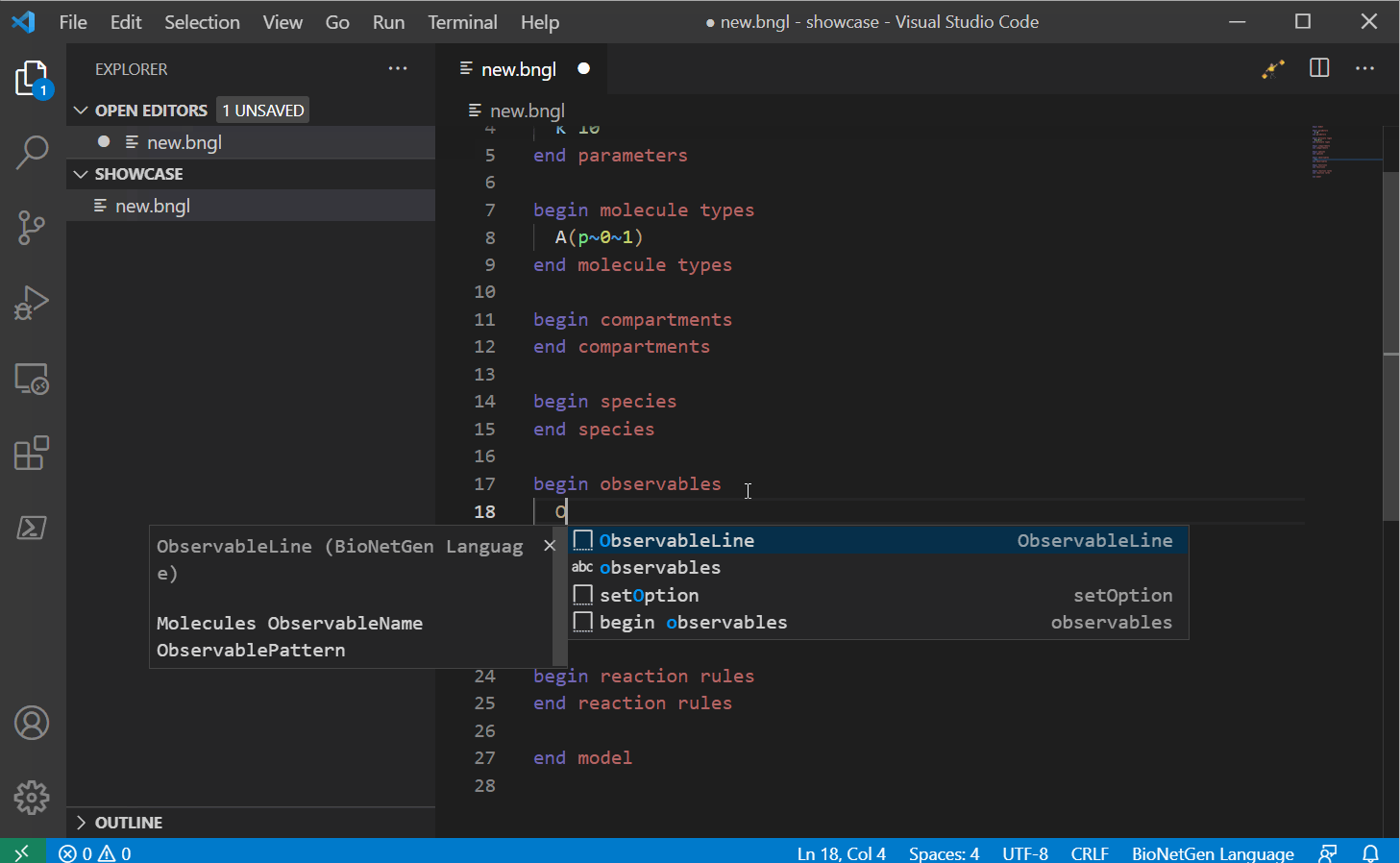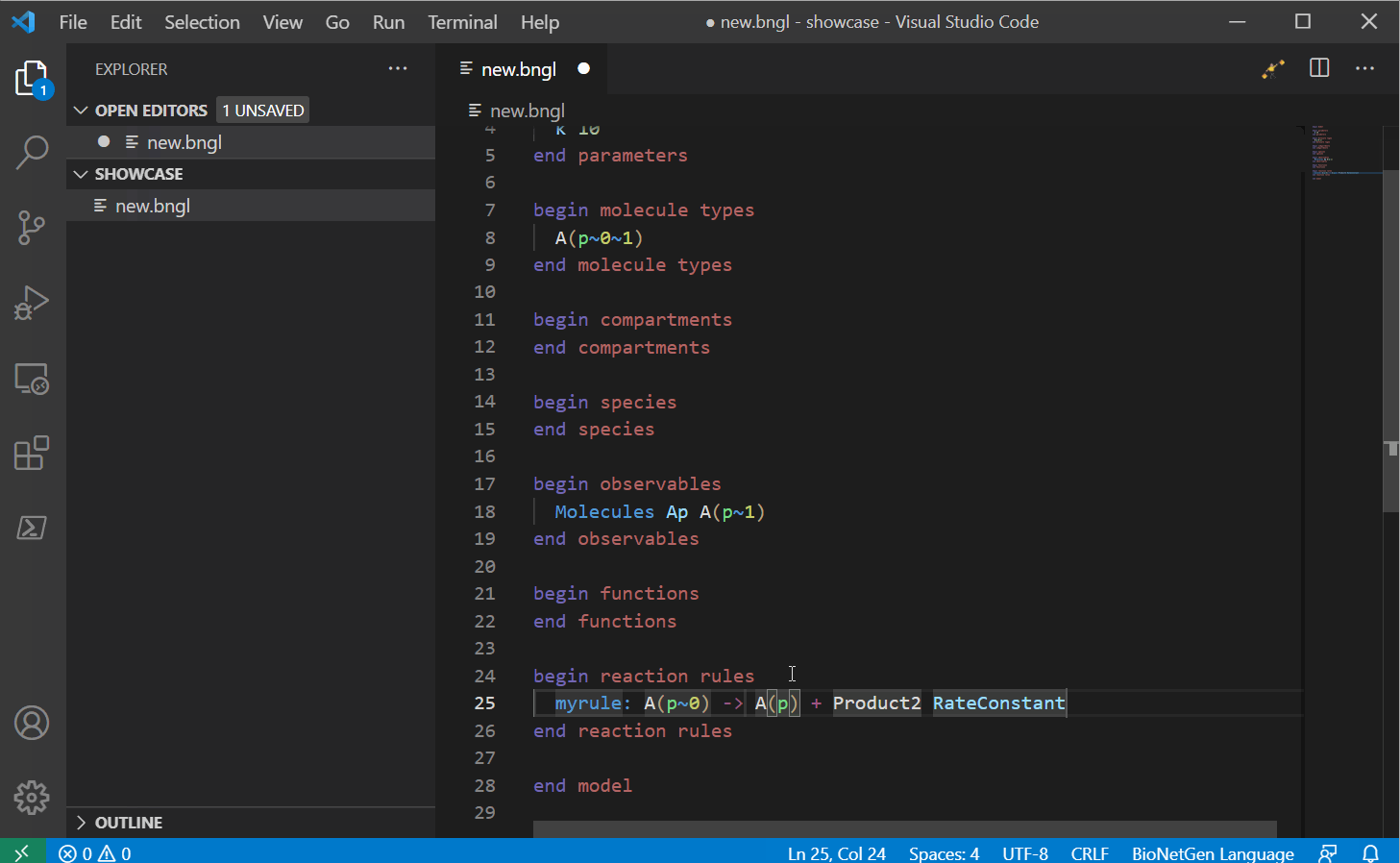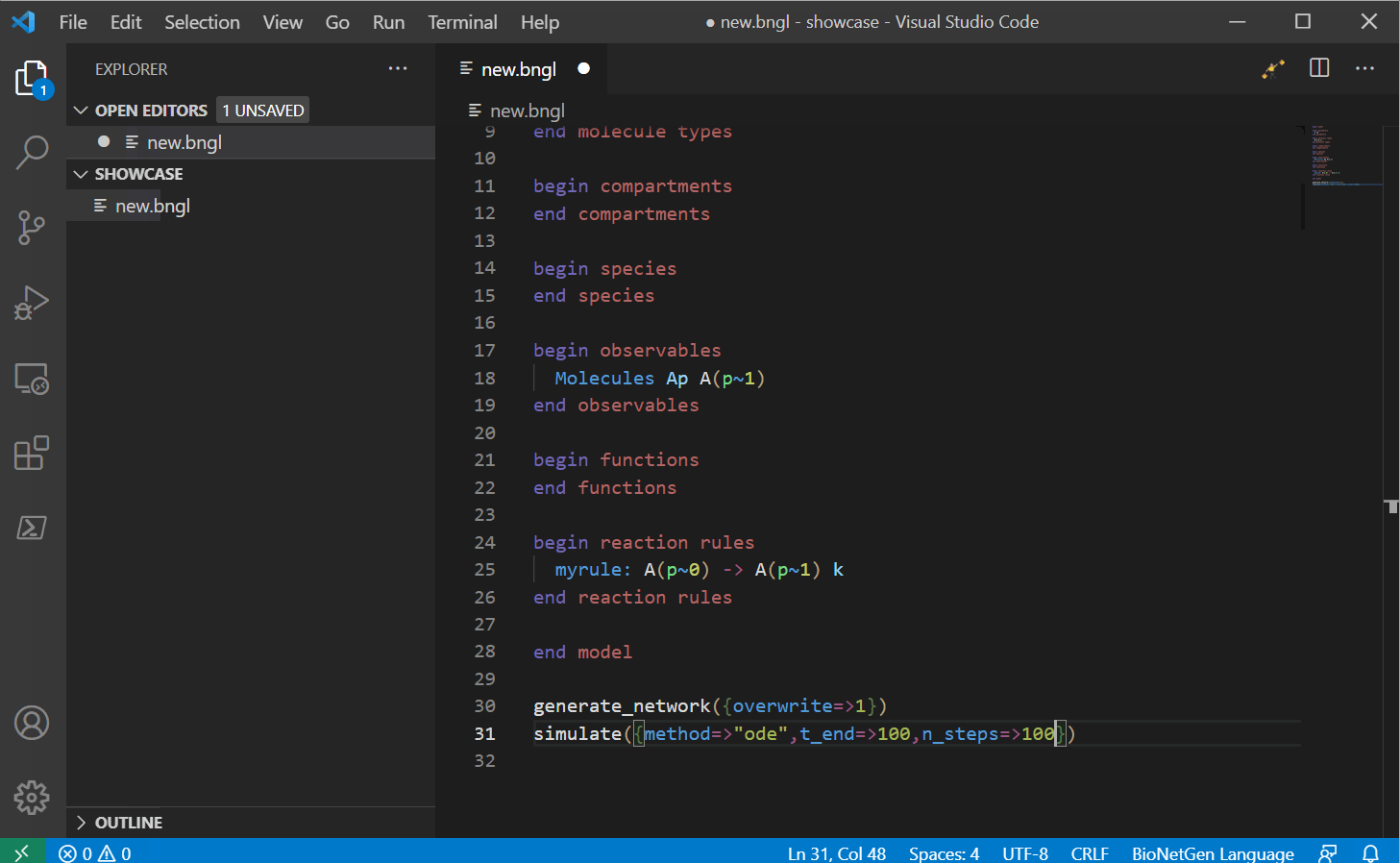
Next you can start writing your model. This VS Code extension supports a large list of snippets that can help you write your model. For a full list, see here, we will update this with a snippet guide in the future.
Using the correct theme
If you notice that there is no highlighting on certain parts of the model or if the colors don’t
match with the figures presented here, please make sure you have the dark-bngl theme
activated (see
here to learn
how to select color themes). Currently only a dark theme is supported, we will include a light
version in the future.
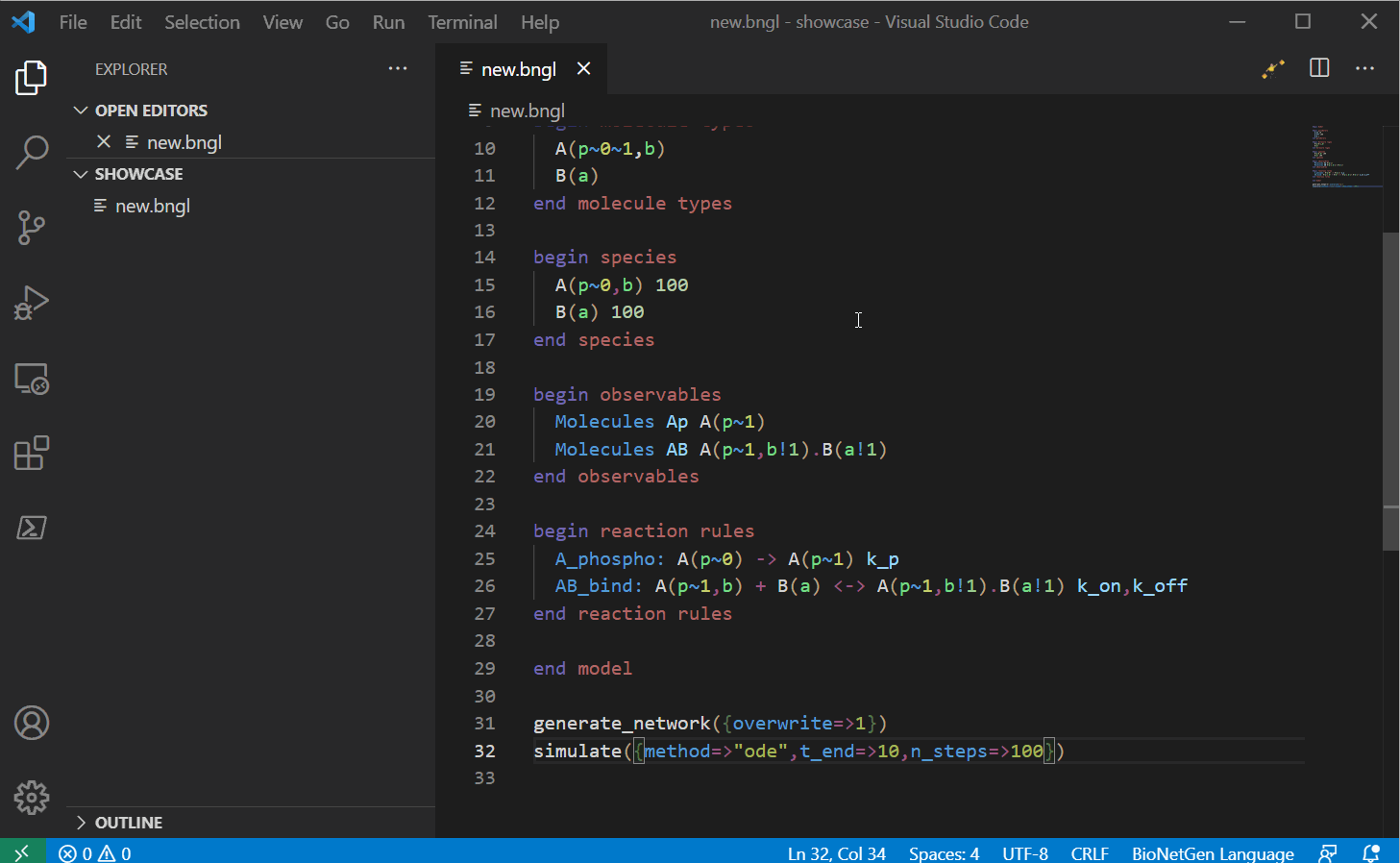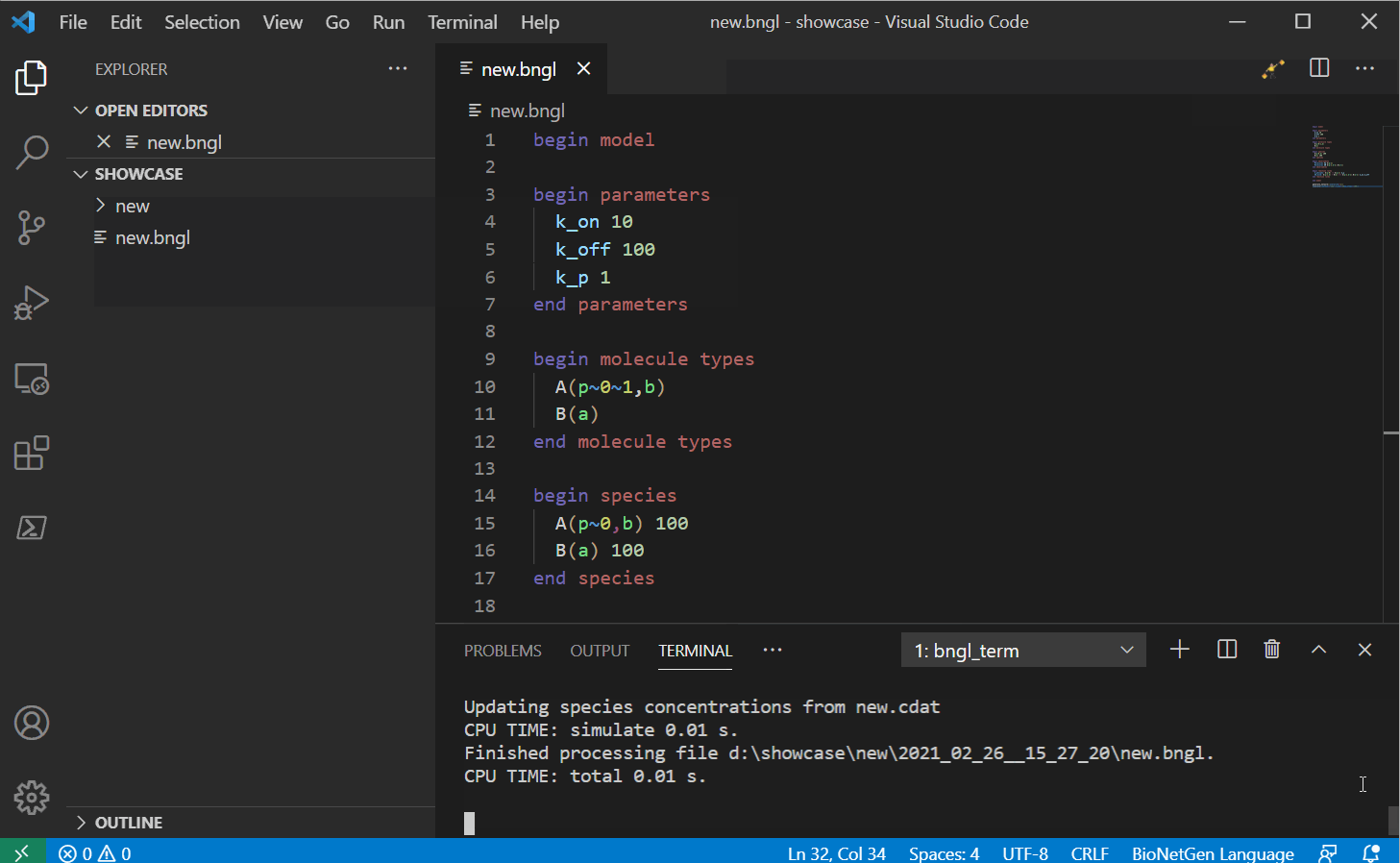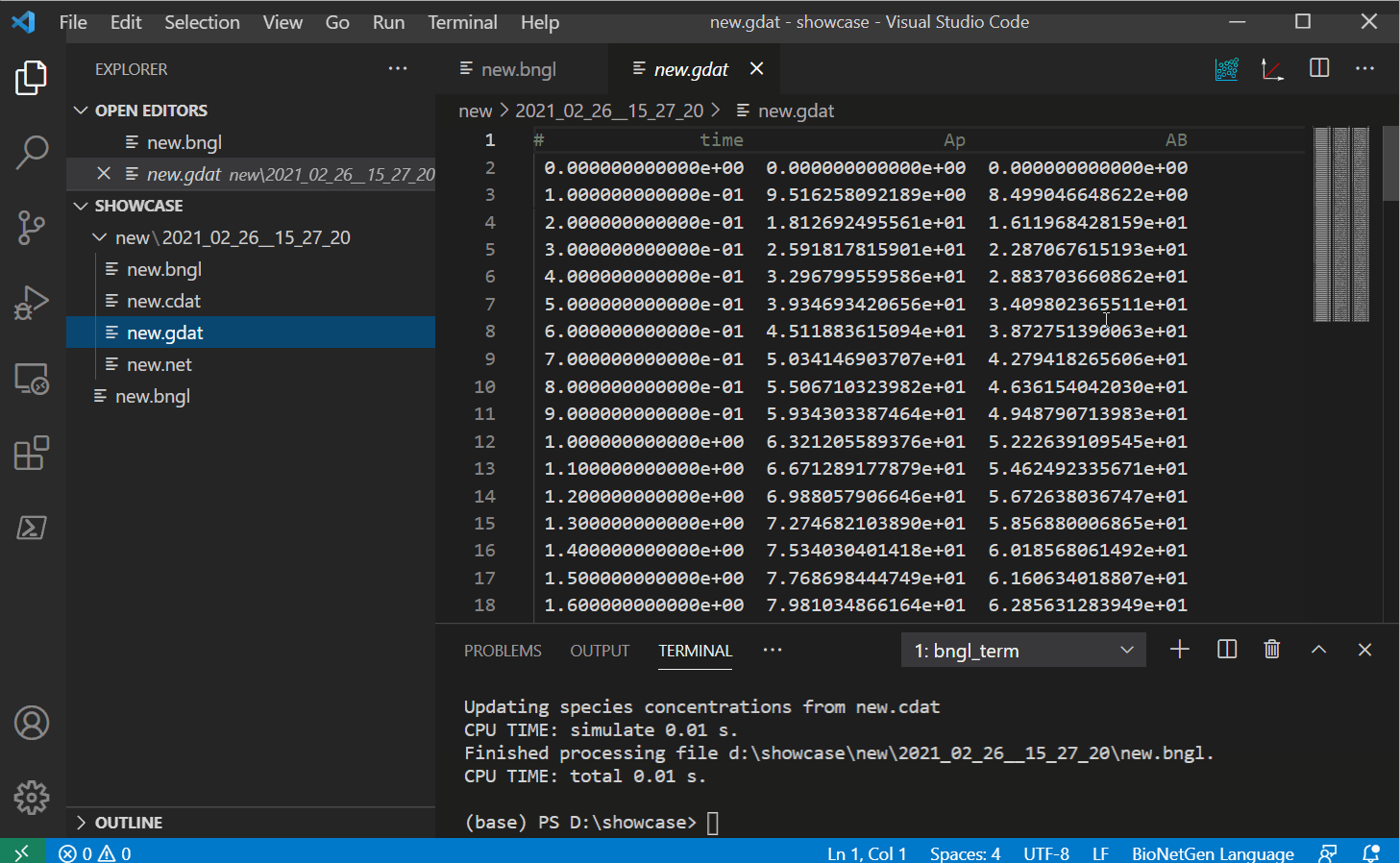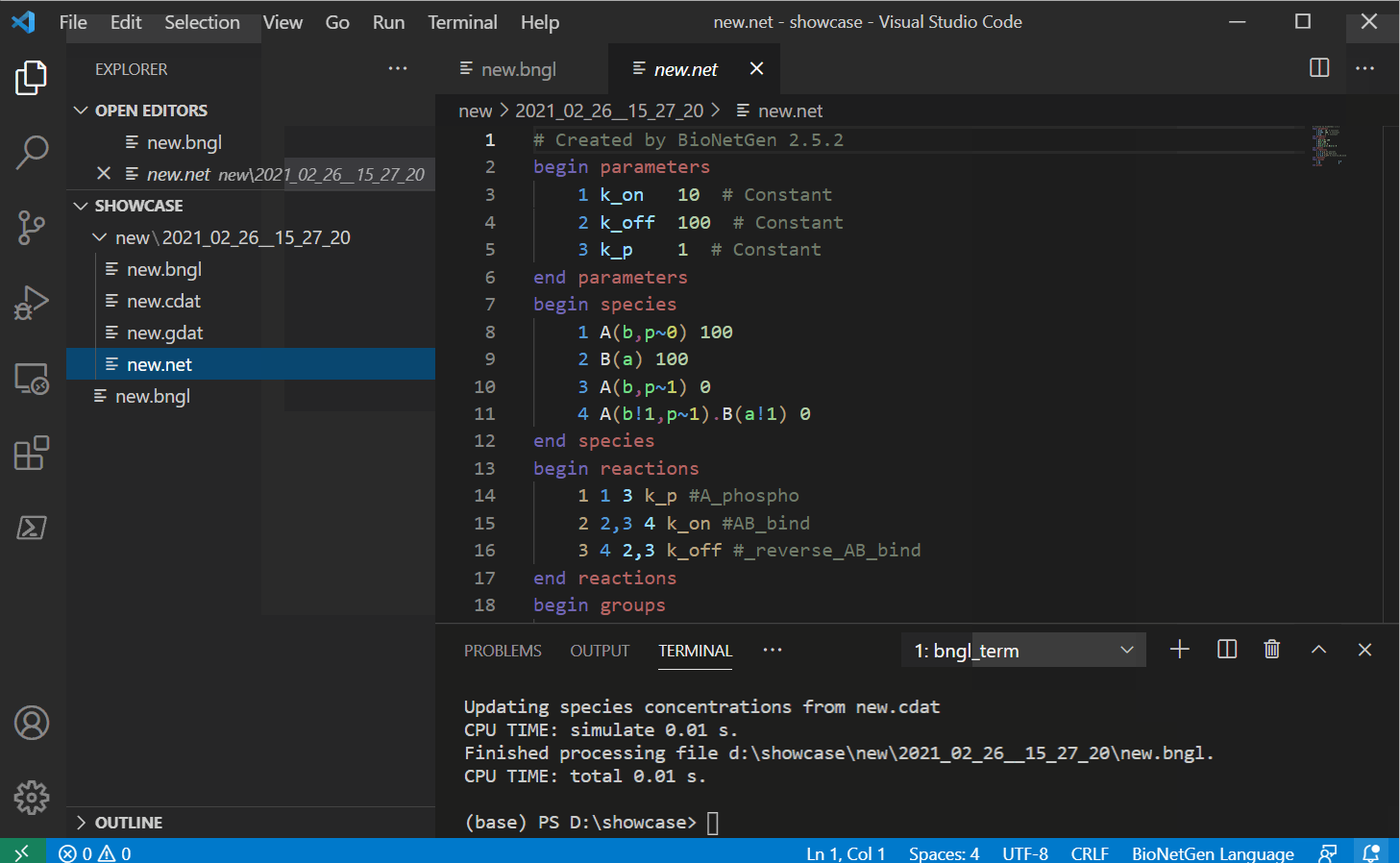
Running a model
Once you finished writing the model, you can try running it. For the run button to work, the
default terminal window VS Code opens should have access to Perl,
Python3 (preferably
anaconda python) and the
PyBioNetGen library. See
Installation for more instructions on how to install the
library. You can test if you have the library correctly installed by
opening a new terminal
and running bionetgen -h.
Once you press the run button (or use the shortcut CTRL/CMD+SHIFT+F1), the extension should
create a new folder with the same name as the model. A time stamped folder will also be created
and the current model will be copied under there and the extension will use the terminal and the
PyBioNetGen library to run the model. Once the run
completes, if the run created a .gdat file, it should open automatically.
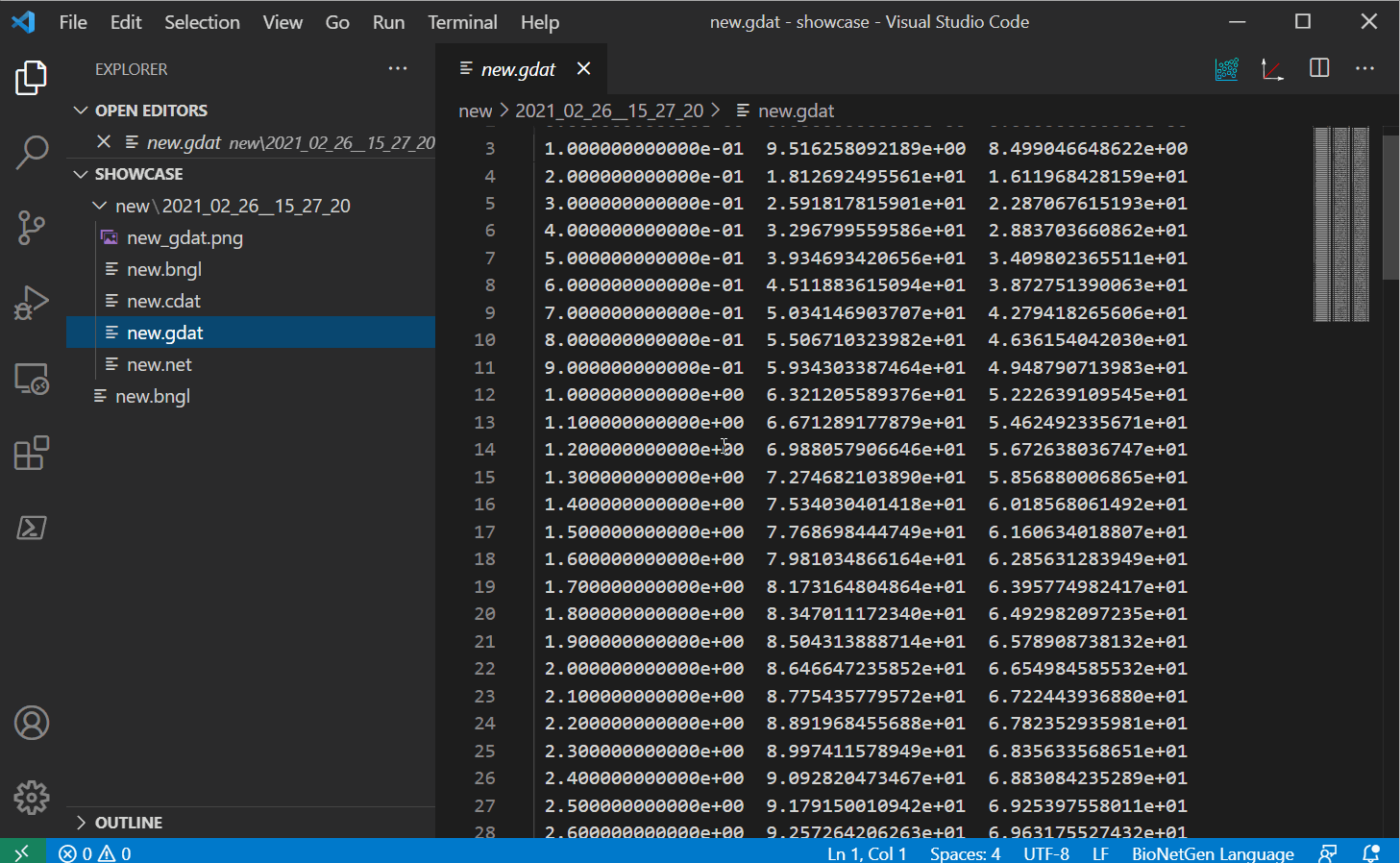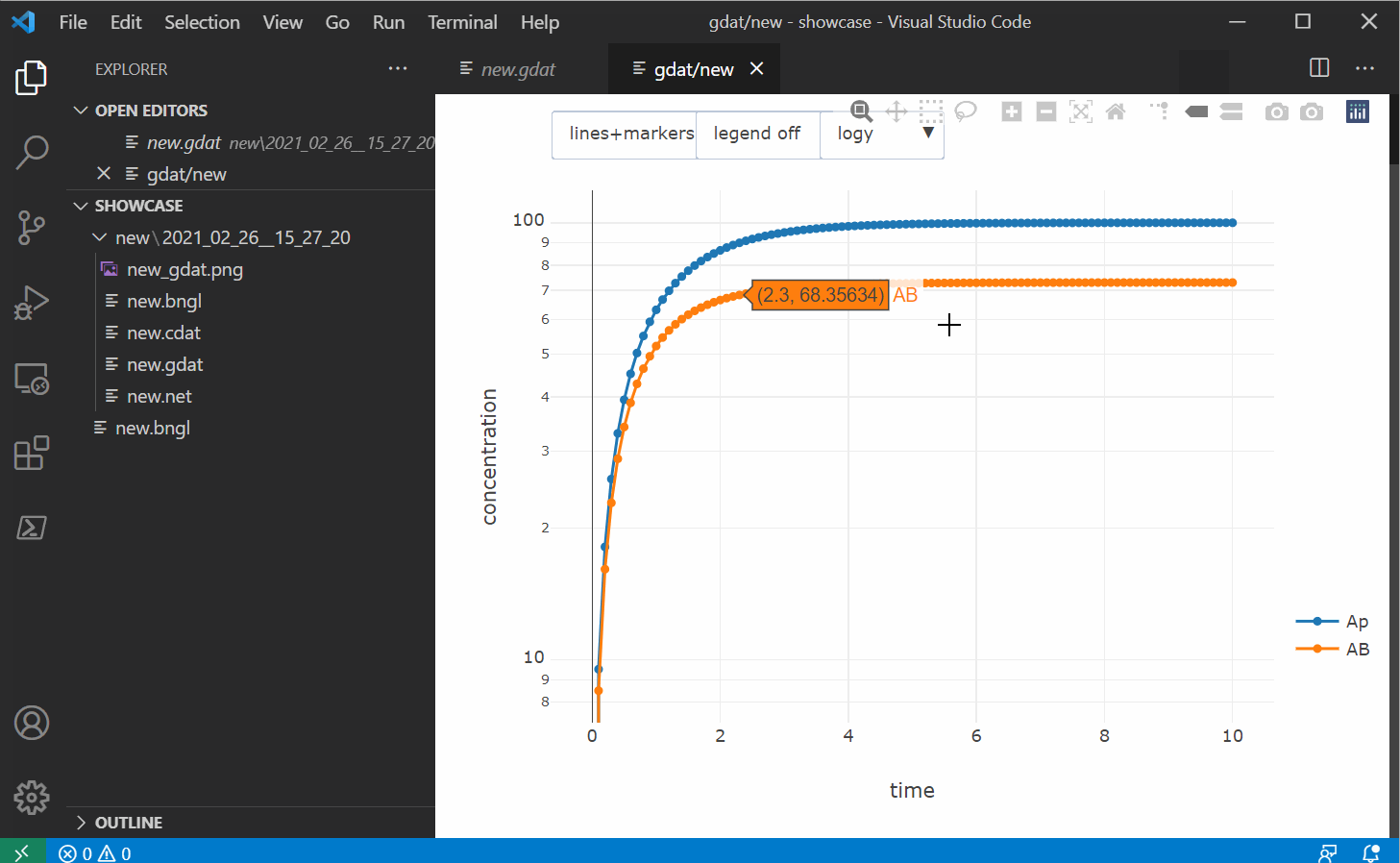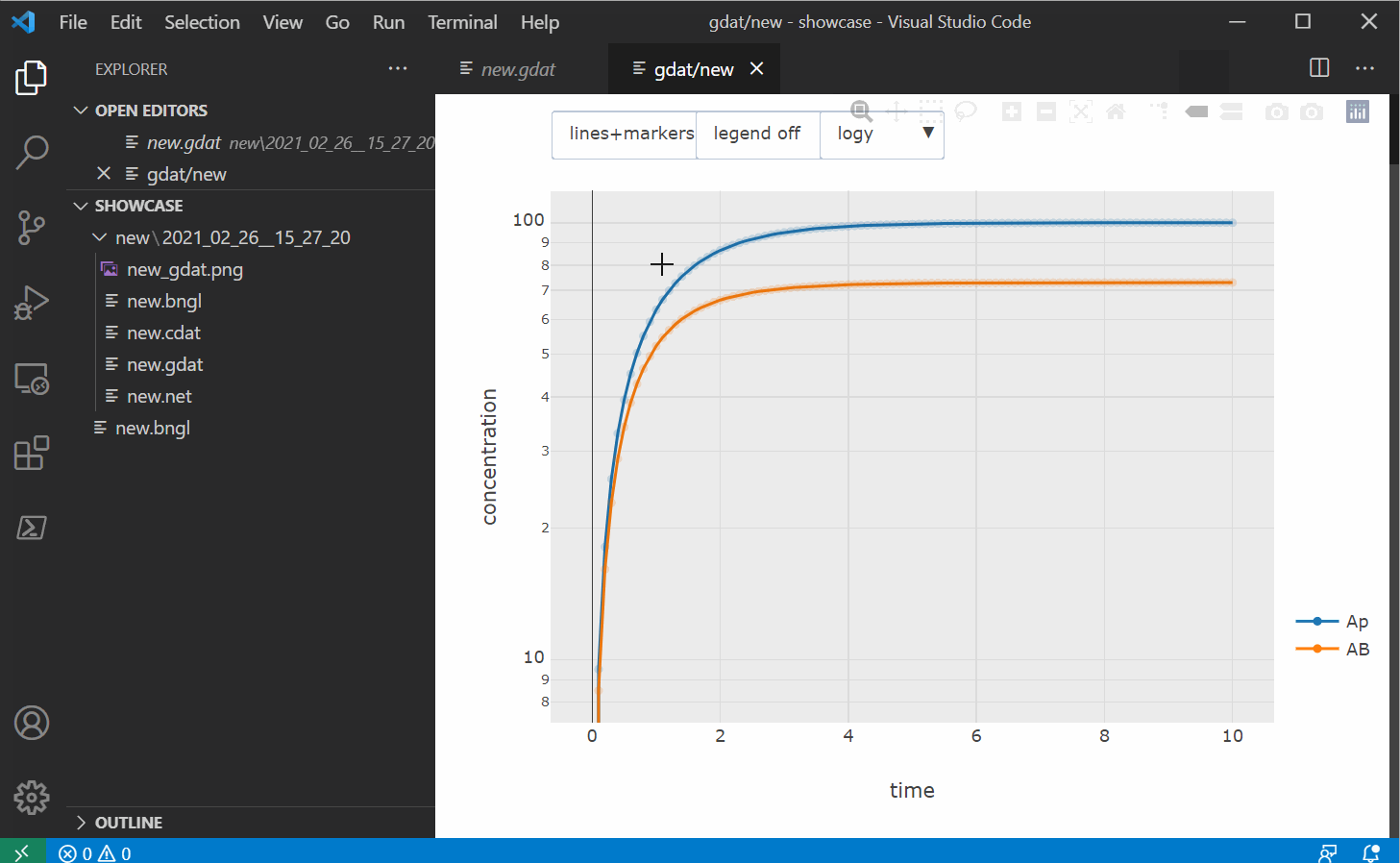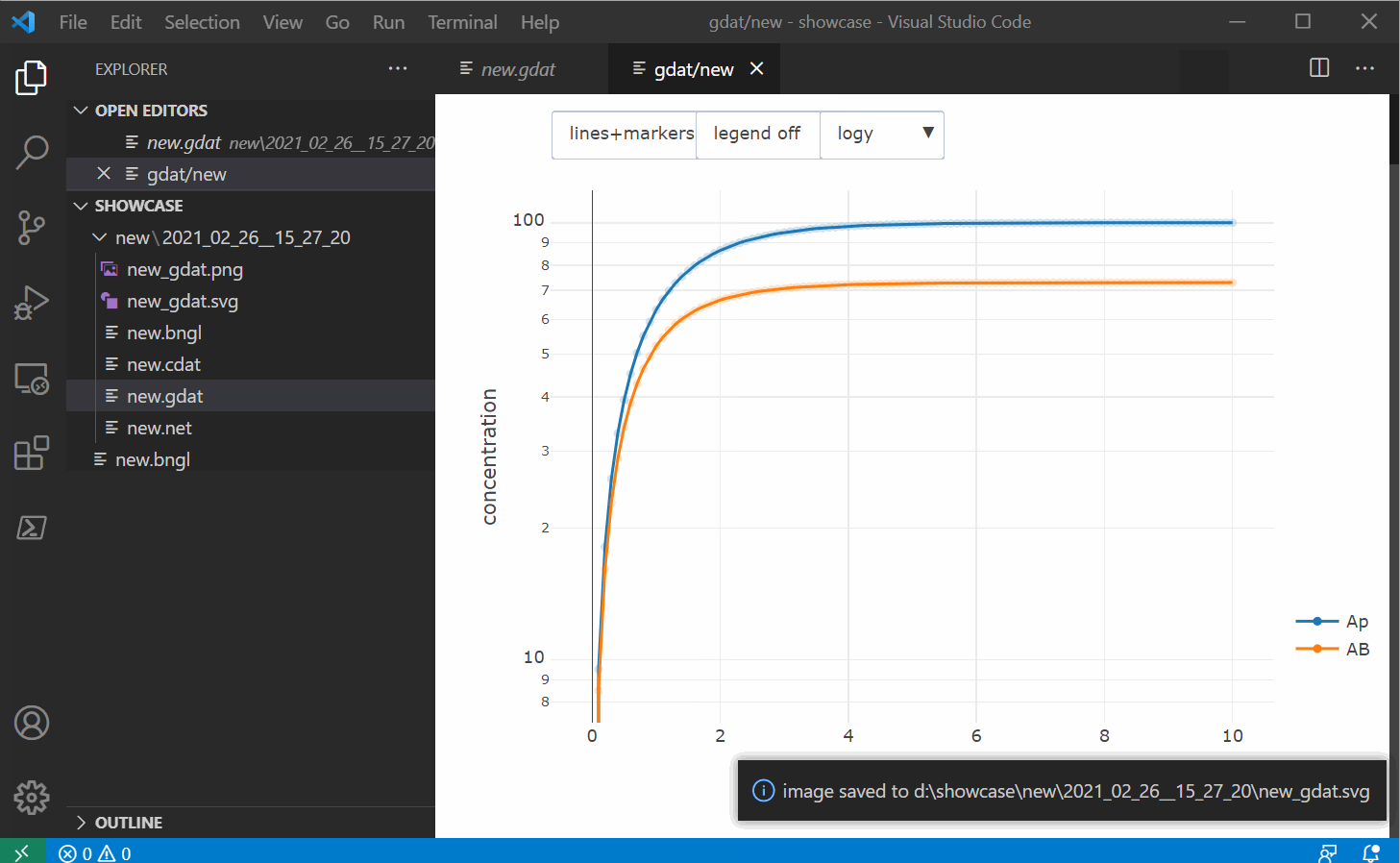
Plotting results
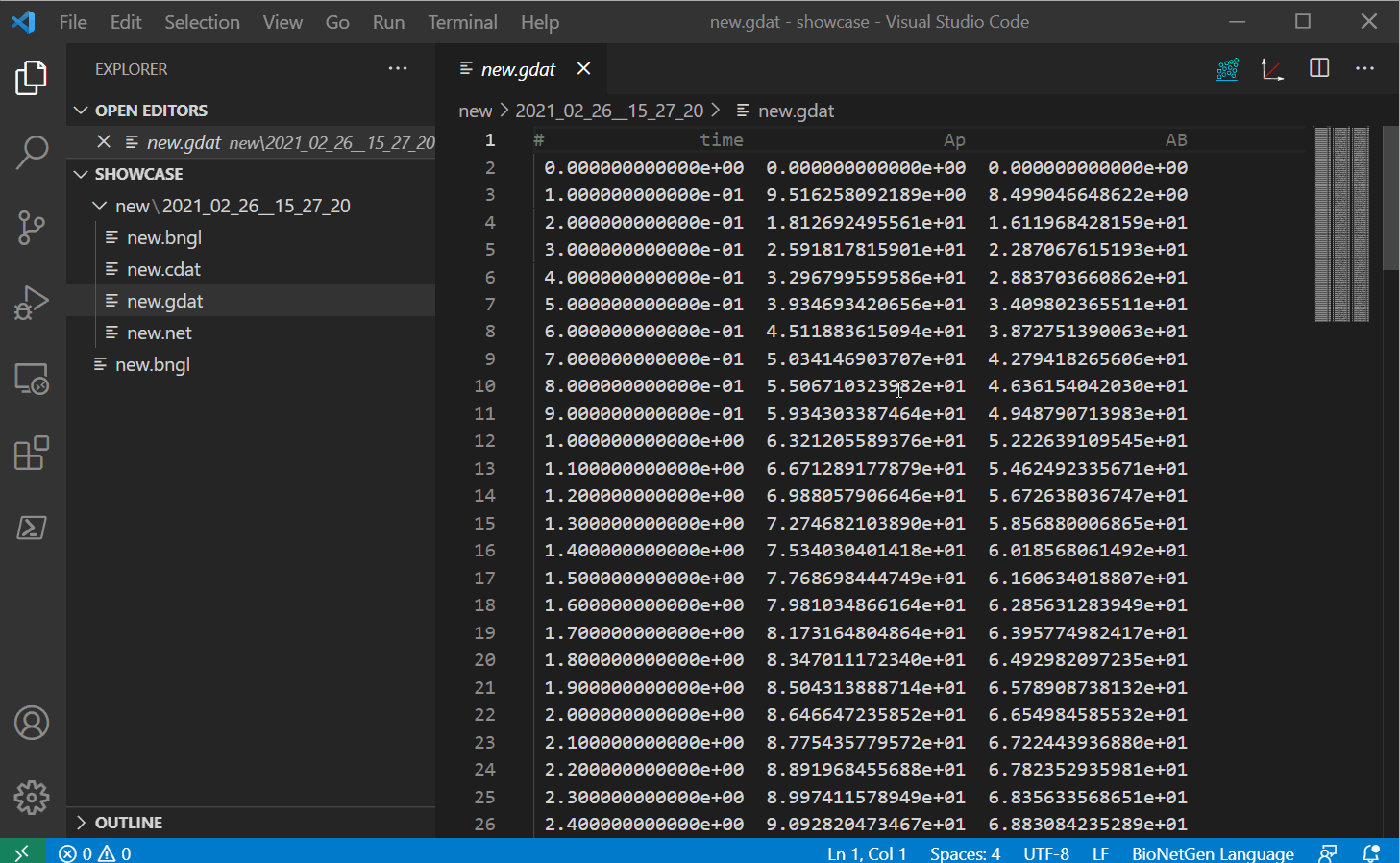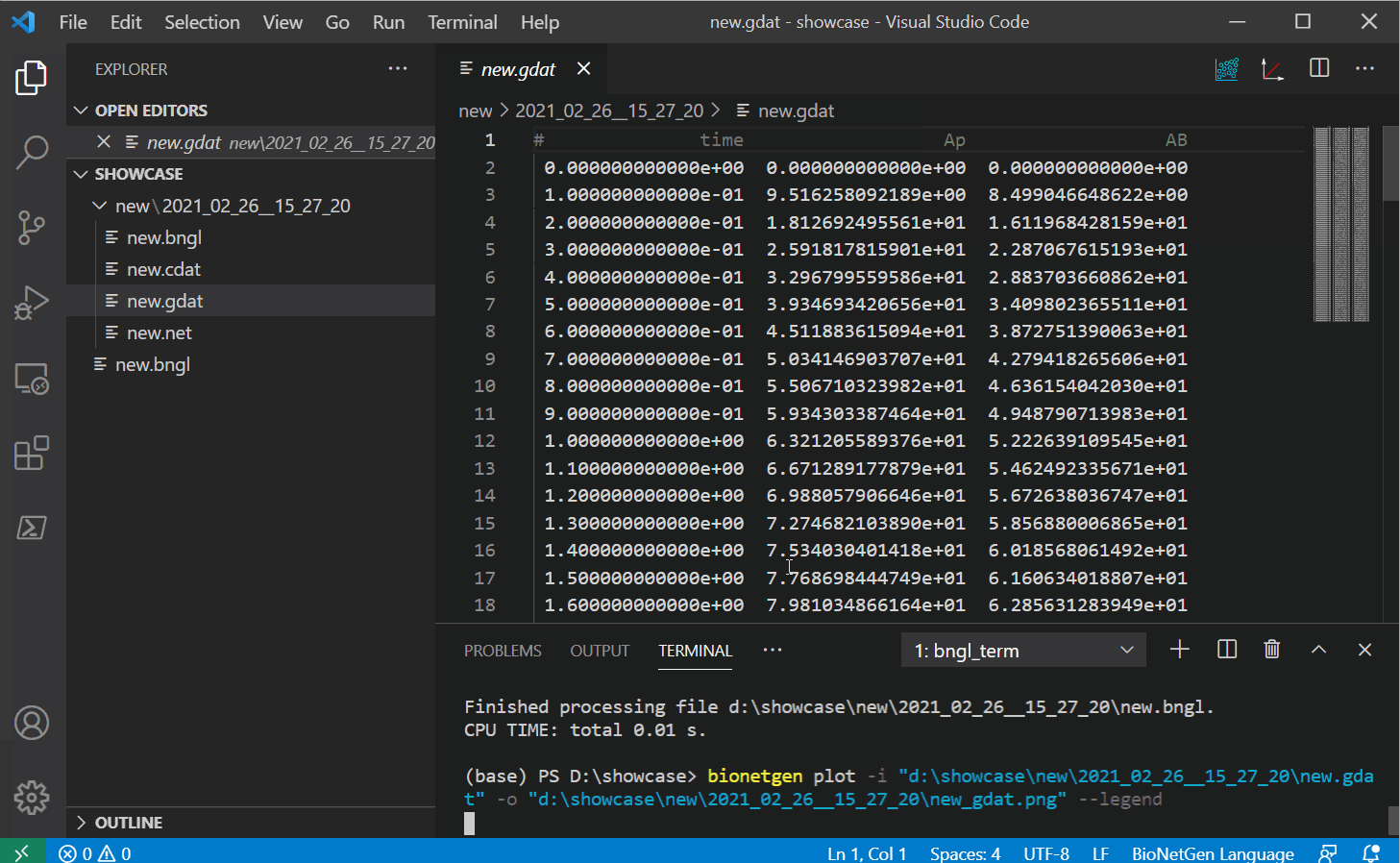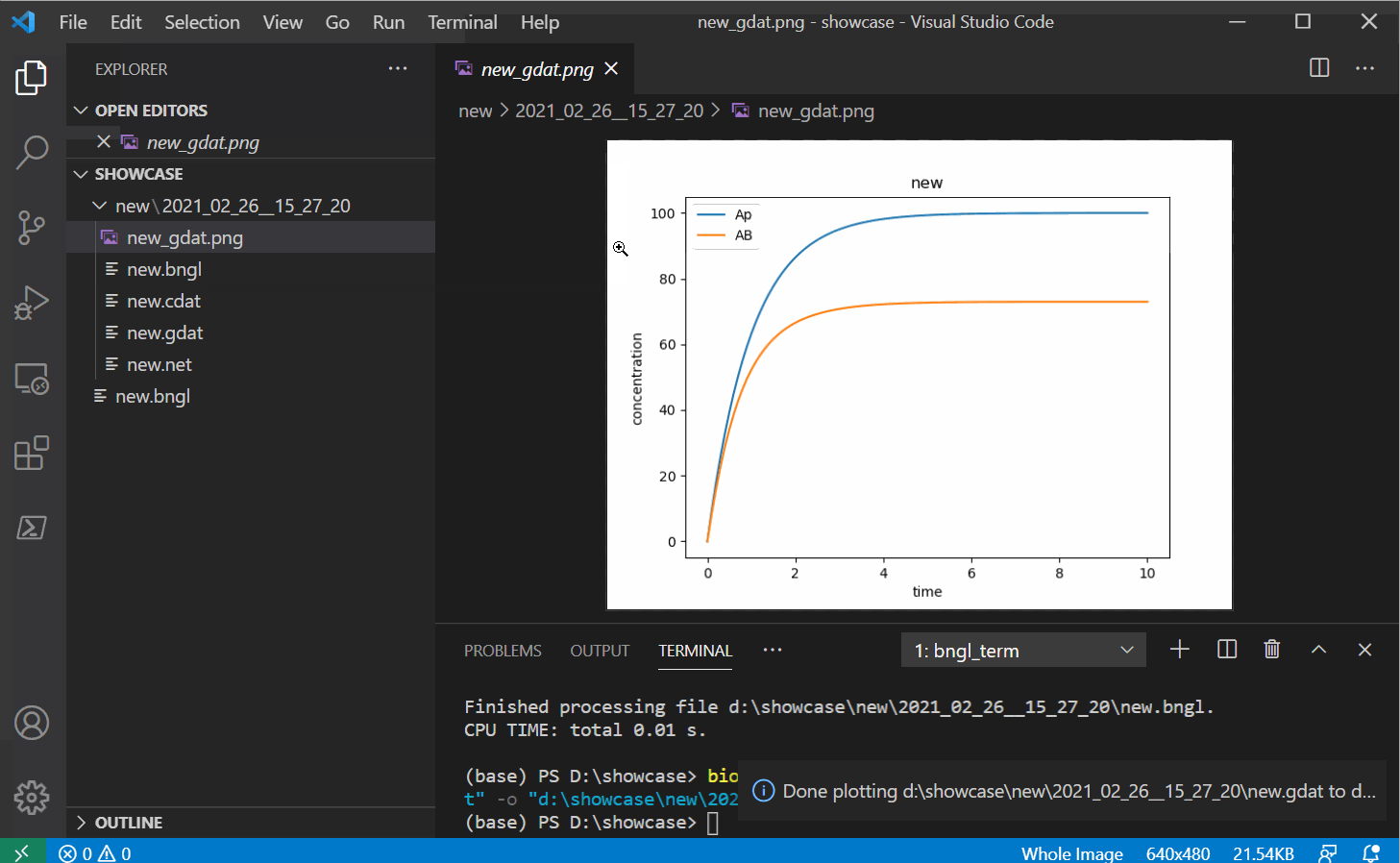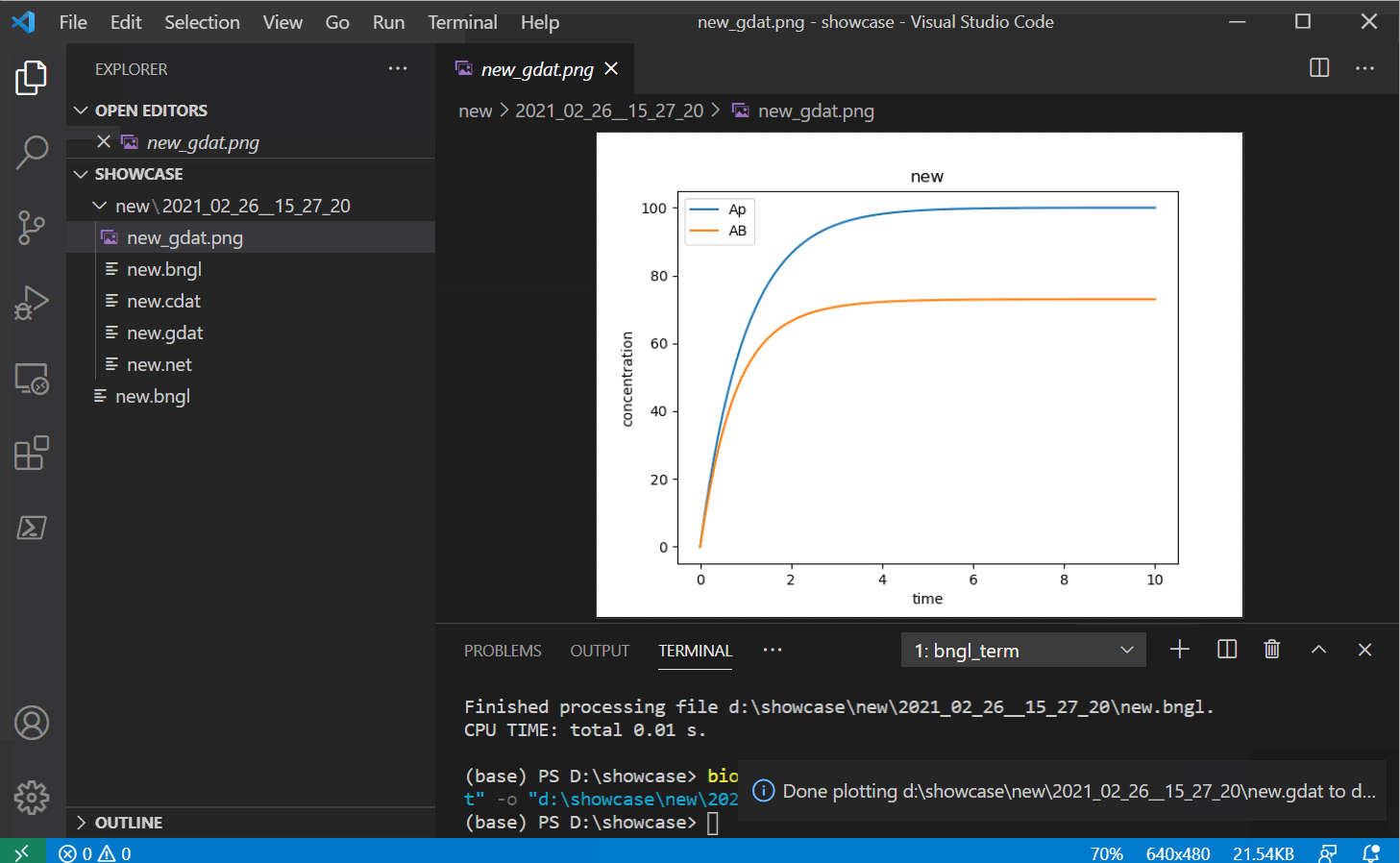
Once you have some gdat/cdat/scan files to look at, you can open one and you should see two
new buttons instead of the run button. The white and red button on the right should plot your
file into a png with some basic defaults (options to change these options will be provided in
future releases).
The blue button on the left should open a new window with an interactive plot (thanks to
plotly.js). You can also use the CTRL/CMD+SHIFT+F1
shortcut. The plotly window also allows you to save the image as a png as well as a svg file and
has interactive features. You can also change the plotting type
to markers or lines+markers and use one of the selection tools to sub-select time series. If you
select a region on the plot with no data points, every time series in the original dataset will
be shown again.
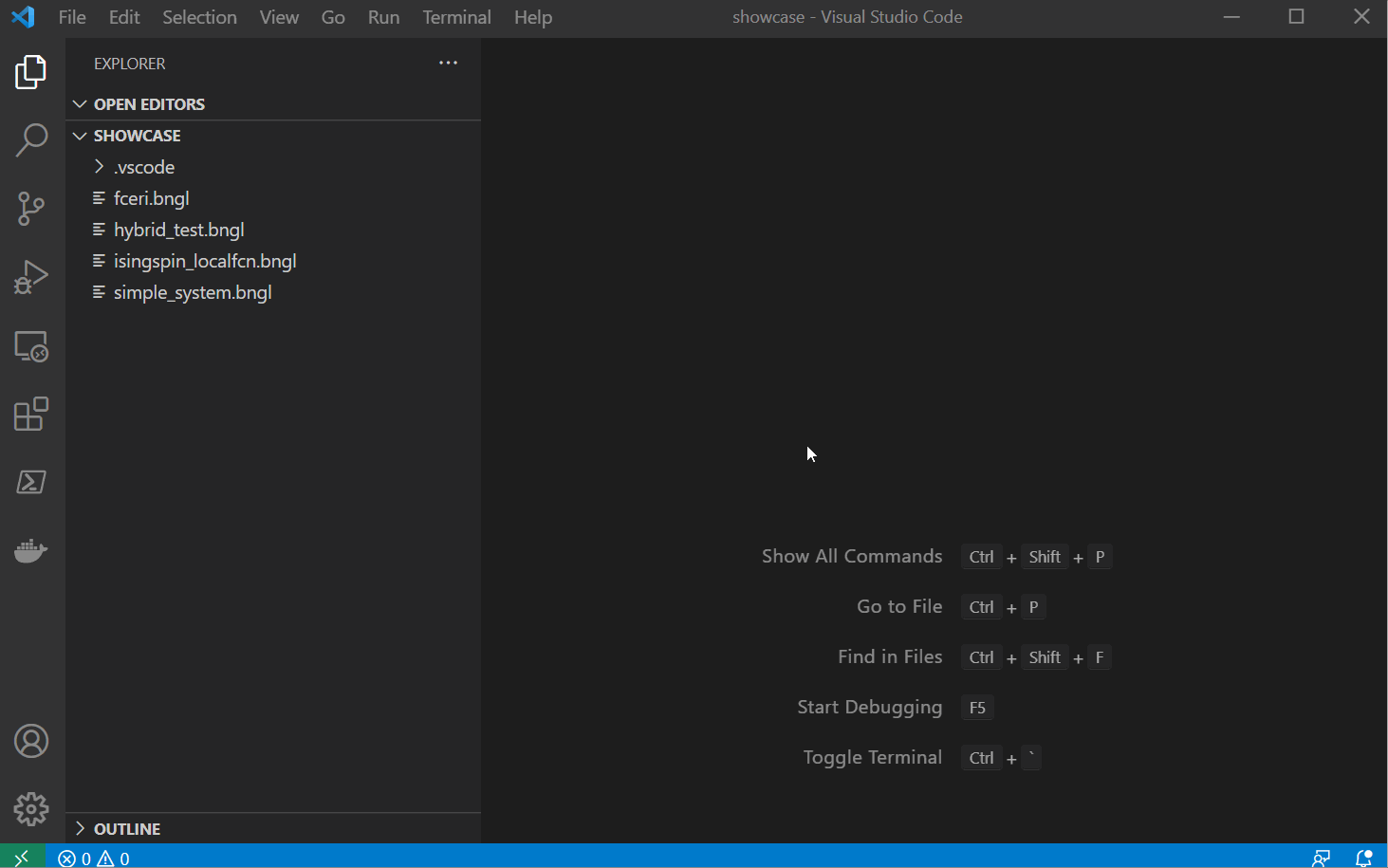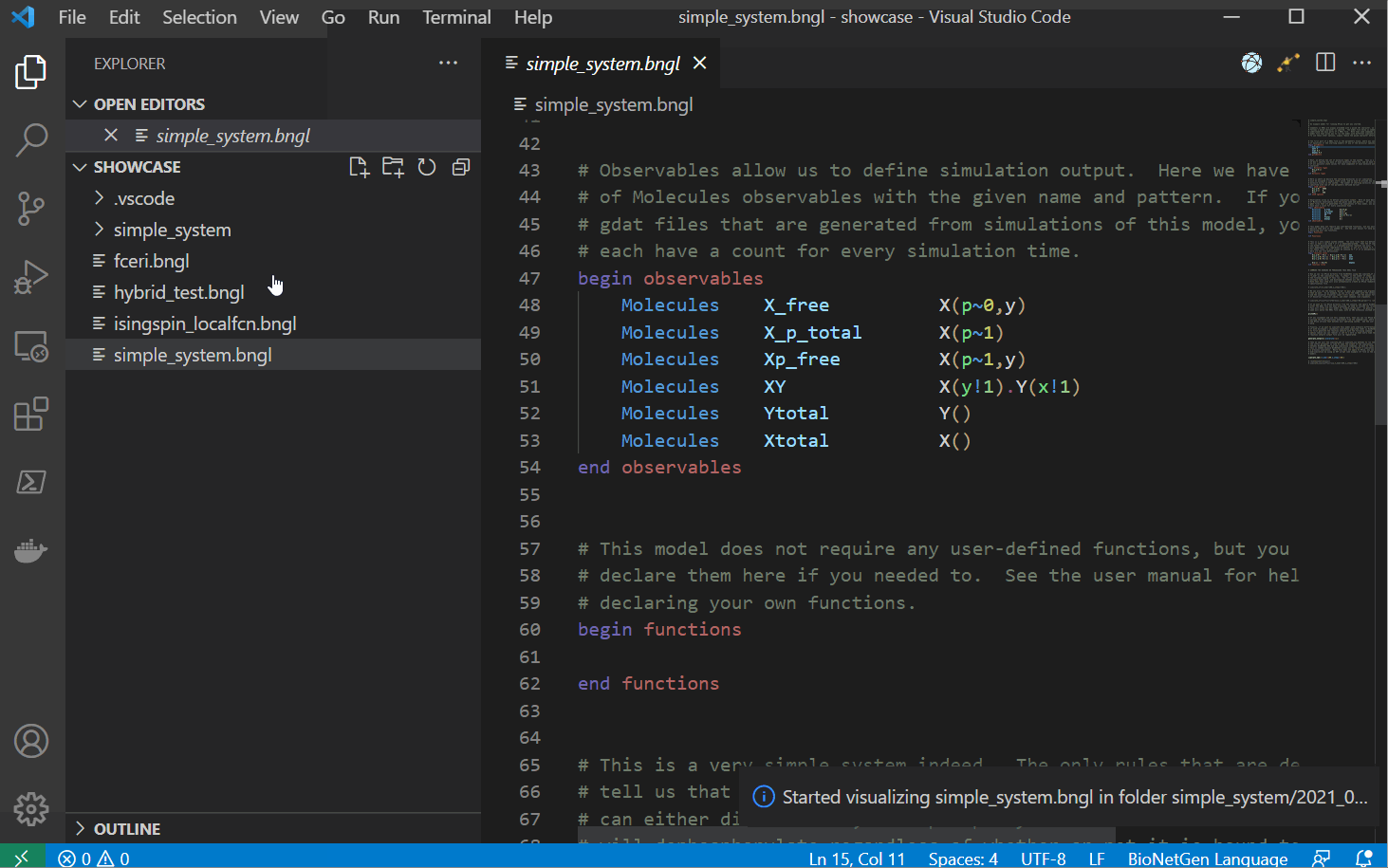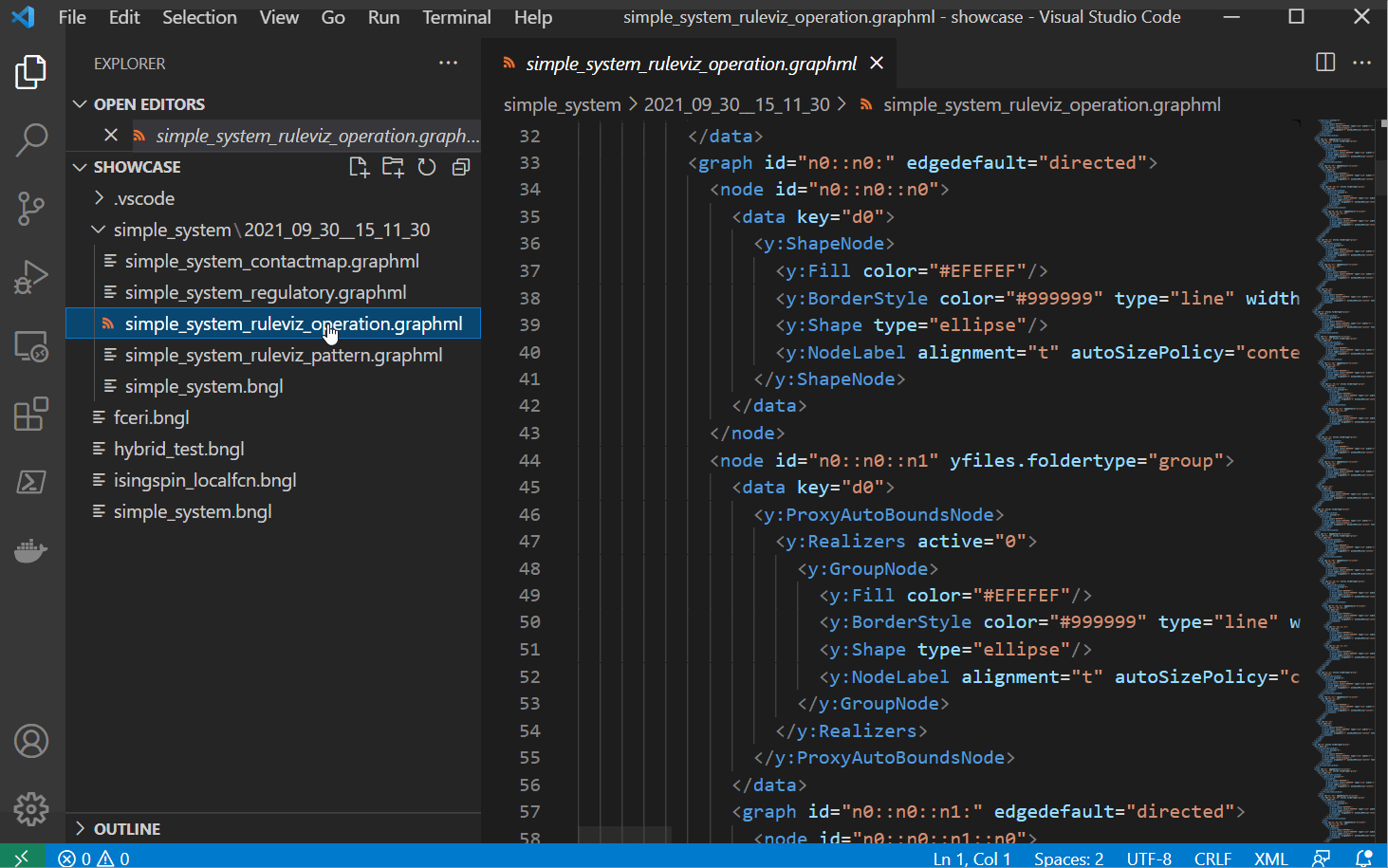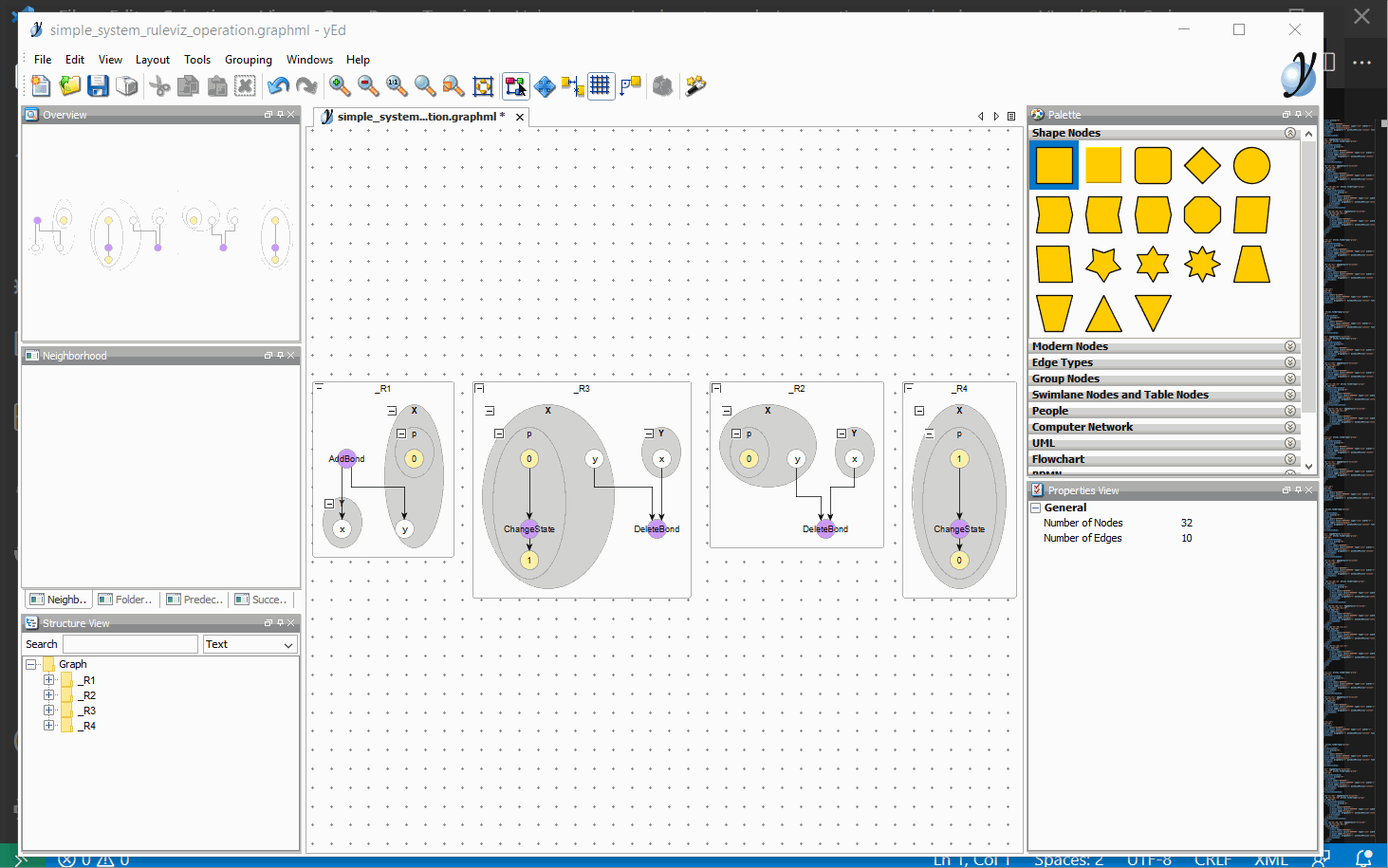
Visualization
You can also click on the visualize button that can be found to the left of the run button. This will generate all possible visualizations that’s available to BioNetGen in a separate folder as GraphML files. These files are designed to be used in conjunction with yEd. If you have yEd installed, you can associate yEd with GraphML files in your OS (example for windows 10). If you also install the open in external app extension for VS Code you can right click the GraphML files and click on Open in external App to open the graph in yEd.