Command Line Interface
The command line tool comes with several subcommands. For each command, you can see the help text with the command
bionetgen subcommand -h
Run
This subcommand simply runs a model:
bionetgen run -i mymodel.bngl -o output_folder
This will run mymodel.bngl under the folder output_folder.
If no output folder is specified, then the temporary folder used while running the subcommand will be deleted upon completion.
Plot
This subcommand allows you to make a simple plot from a gdat/cdat or scan file:
bionetgen plot -i mymodel.gdat -o gdat_plot.png
You can see all the available options by running bionetgen plot -h
optional arguments:
-h, --help show this help message and exit
-i INPUT, --input INPUT
Path to .gdat/.cdat file to use plot
-o OUTPUT, --output OUTPUT
Optional path for the plot (default:
"$model_name.png")
--legend To plot the legend or not (default: False)
--xmin XMIN x-axis minimum (default: determined from data)
--xmax XMAX x-axis maximum (default: determined from data)
--ymin YMIN y-axis minimum (default: determined from data)
--ymax YMAX y-axis maximum (default: determined from data)
--xlabel XLABEL x-axis label (default: time)
--ylabel YLABEL y-axis label (default: concentration)
--title TITLE title of plot (default: determined from input file)
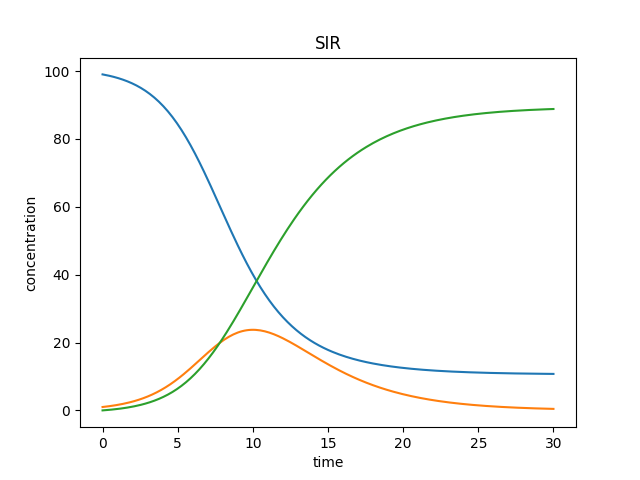
Resulting plots should look similar to this:
Visualize
This subcommand creates .graphml files to be used by an external graph editor (yEd) for model visualization, including contact maps.
bionetgen visualize -i mymodel.bngl -o output_folder
You can see all the available options by running bionetgen visualize -h
optional arguments:
-h, --help show this help message and exit
-i INPUT, --input INPUT
Path to BNGL model to visualize
-o OUTPUT, --output OUTPUT
(optional) Output folder, defaults to current folder
-t TYPE, --type TYPE (optional) Type of visualization requested. Valid options are: 'ruleviz_pattern','ruleviz_operation', 'contactmap', 'regulatory' and
'atom_rule'. Regulatory and atom rule graphs are the same thing, defaults to 'contactmap'.
Notebook
This subcommand is in its early stages of development. The subcommand is used to generate a simple Jupyter notebook. You can also give your model as an argument and the resulting notebook will be ready to load in your model using PyBioNetGen library.
bionetgen notebook -i mymodel.bngl -o mynotebook.ipynb
Info
This subcommand simply prints out information about software versions and installation paths.
bionetgen info
Atomize
The CLI includes one more subcommand, atomize, which is detailed further in Atomizer.
Tutorial
For a brief tutorial showing how to use the CLI on a simple BNGL model, please see CLI Tutorial.